
From Fold to Function: Inside Boltz
In this series: From AlphaFold2 to Boltz-2: The Protein Prediction Revolution
- Part 1: Deconstructing the Fold with AlphaFold2
- Part 2: Denoising the Complex with AlphaFold 3
- Part 3: From Fold to Function: Inside Boltz (You are here)
- Part 4: Appendix (From Alphafold 2 to Boltz-2)
From Co-Folding to Co-Design: The Definitive Deep Dive into the Boltz-1 and Boltz-2 Models
Introduction
Getting the pose right isn’t always enough—just ask patients who saw their treatments fail overnight.
In the late 2000s, cancer patients treated with the revolutionary drug imatinib (Gleevec) faced sudden relapses. Investigations traced the culprit to a single mutation, T315I, in the BCR-ABL protein. X-ray crystallography showed that imatinib still bound snugly into its usual pocket, but this subtle atomic swap completely disrupted crucial hydrogen bonds, dropping binding affinity by several orders of magnitude and rendering the drug clinically ineffective.This devastating scenario underscores a crucial lesson: predicting the right structure doesn’t guarantee a drug will remain effective—it’s equally vital to understand how tightly a molecule binds to its target.
In Part 1 of this series [1], we explored how AlphaFold 2 cracked biology’s 50-year-old protein-folding challenge. In Part 2 [2], we watched AlphaFold 3 expand this revolution to model entire biomolecular assemblies, though at the time, access was restricted by licensing[3].
The landscape shifted dramatically in late 2024 with the arrival of Boltz-1 [4], the first fully open-source model to deliver AlphaFold 3-class predictions freely to academia and industry alike. Within weeks, biotech firms integrated Boltz-1 into their drug discovery pipelines, signaling the demand for open, powerful structure prediction.
Yet Boltz-1 was only the beginning. In mid-2025, its successor, Boltz-2 [5], moved beyond simply predicting structural fit to addressing the deeper question of molecular affinity—how tightly two molecules grip one another. Combining diffusion-based co-folding with an innovative affinity head, Boltz-2 achieved binding affinity predictions approaching the gold-standard accuracy of Free-Energy Perturbation (FEP) simulations, but at a computational speed roughly 1000 times faster [6,7].
This post tells the story of Boltz-1 and Boltz-2 as open-source milestones, showcasing how the community evolved the field from merely predicting structures to rapidly and accurately estimating molecular interactions. Together, these models represent a bold leap toward AI-driven molecular co-design, opening new doors in drug discovery and beyond.
🧠 Refresher: Key Concepts from AlphaFold 3
- A Universal Language (Tokens)
AlphaFold 3 swapped its protein‑only view for a token system that covers any molecule:- Biopolymers (proteins, DNA/RNA) → one token per residue
- Diverse ligands → one token per heavy atom
- The Pairformer & Dethroning the MSA
- Pairformer replaces Evoformer
- MSA offers a quick hint, then steps aside; the 48 cycles focus on geometry
- Triangle Attention still enforces global 3‑D consistency
- A Conditional Generative Core (Diffusion)
- Input: a random cloud of atoms
- Guidance: single & pair representations from the Pairformer (the “conditional” blueprint)
- Output: a coherent, physically plausible structure achieved step‑by‑step
Boltz starts from this blueprint, then adds steering potentials and an affinity head to shift the focus from fit to tightness.
Boltz-1 is an open-source reimplementation of AlphaFold 3 that predicts the 3D structure of protein complexes (and protein–ligand interactions) with AlphaFold3-level accuracy. It adopts AlphaFold 3’s two-stage strategy: a Pairformer trunk that builds a detailed geometric blueprint of all interacting molecules, and a diffusion-based generative module that co-folds the complex. Boltz-1 introduced efficiency improvements (like TriFast fused attention kernels) and made multiple small architectural tweaks for stability and logic, all while being freely available under an MIT license. The open release of Boltz-1 has dramatically lowered barriers to applying state-of-the-art structure prediction in practice.
Boltz-2 goes a step further, transforming the co-folding model into a co-design model. It adds a suite of physics-inspired “steering” potentials that gently guide the generative process to produce physically realistic models (no more distorted bond angles or left-handed amino acids). It also gives users more control—you can bias the model with known structures (templates), enforce custom distance restraints, or even tell it to mimic experimental conditions (e.g.,
--method cryo-EM for softer, more flexible outputs). Most remarkably, Boltz-2 introduces an affinity prediction head that estimates how strongly a ligand will bind its protein target. This head was trained on over a million experimental binding measurements and approaches the accuracy of gold-standard free energy calculations (which take days) in mere seconds. In short, Boltz-2 doesn’t just predict how molecules will fit together—it predicts how tightly they’ll hold on, bringing us closer to AI-driven drug design.
In traditional co-folding, an AI model predicts how multiple pieces (e.g., two proteins, or a protein and a drug) come together into a complex structure — much like solving a 3D puzzle. With Boltz-2, we're elevating to co-design: not only does the model assemble the puzzle, it also evaluates how good the fit is. This is akin to not just drawing two jigsaw pieces that lock together, but also estimating how strong that lock is. In practical terms, co-design means the AI helps suggest which molecules might work best together, not just how they might look together.
The Foundational Architecture: A Better, Faster, and More Open Engine
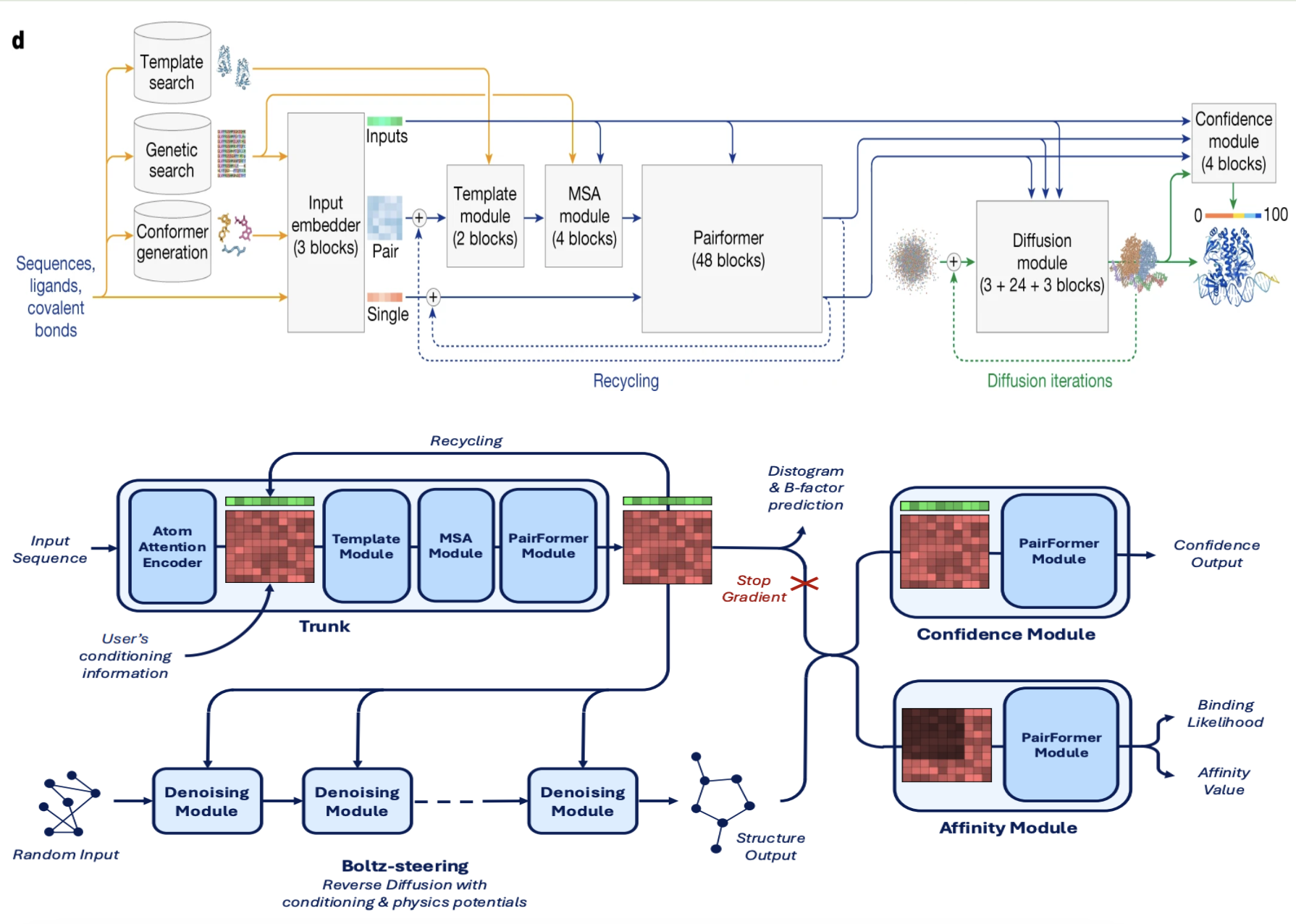 Figure 1. Comparison of AlphaFold 3 (top) and Boltz-2 (bottom). Top: AlphaFold 3 trunk‑to‑diffusion pipeline, redrawn from Abramson et al. 2024 [8]. Bottom: Boltz‑2 pipeline, adapted from Passaro et al. 2025 [5]. Boltz‑2 deepens the trunk (64 vs 48 Pairformer blocks), swaps in TriFast kernels for speed, and adds two new heads: a lightweight confidence module and a specialised affinity module that approaches free‑energy‑perturbation accuracy—all within the same network.
Figure 1. Comparison of AlphaFold 3 (top) and Boltz-2 (bottom). Top: AlphaFold 3 trunk‑to‑diffusion pipeline, redrawn from Abramson et al. 2024 [8]. Bottom: Boltz‑2 pipeline, adapted from Passaro et al. 2025 [5]. Boltz‑2 deepens the trunk (64 vs 48 Pairformer blocks), swaps in TriFast kernels for speed, and adds two new heads: a lightweight confidence module and a specialised affinity module that approaches free‑energy‑perturbation accuracy—all within the same network.
At its core, Boltz-1 adopted the powerful blueprint pioneered by AlphaFold 3 (see Part 2 [2]): a deep learning system composed of a Pairformer trunk to learn rich representations of the entire molecular system, and a diffusion-based generator to translate those representations into a 3D structure, Figure 1. The guiding philosophy was clear: match the performance of the closed-source model, but make it faster, more efficient, and accessible to all.
Speaking the Universal Language of Molecules
Boltz inherits AlphaFold 3’s token trick: For standard biomolecules (proteins, DNA, RNA), each building block (residue or nucleotide) is represented as a single token, holding idealized geometry from an internal reference library. For diverse, non-standard molecules like drug ligands, each heavy atom becomes its own token, initialized with a realistic 3D conformer generated by tools such as RDKit9.
This token-based approach allows Boltz to seamlessly handle chemically diverse molecular scenes. Each token’s initial atomic features are first refined through a quick atom-level transformer (the “Input Embedder”), preparing a rich set of molecular representations that feed directly into the Pairformer.
The Pairformer Trunk: A Deeper, Faster Engine for Molecular Reasoning
The computational “brain” of Boltz is the Pairformer—the module first introduced in AlphaFold 3 as a successor to the Evoformer. While the Evoformer devoted much of its attention to evolutionary data (MSAs), the Pairformer sharply reduces this dependency, focusing instead on directly refining geometric relationships between every pair of molecular tokens.
In Boltz-1, the Pairformer trunk closely mirrored AlphaFold 3’s architecture, stacking 48 transformer layers to iteratively enhance its pairwise geometric understanding. Boltz-2, however, takes this further by expanding the trunk to 64 layers. Typically, adding depth in this way brings a steep computational cost—but Boltz-2 introduces a key optimization.
The main computational bottleneck for the Pairformer lies in its triangular attention operations. As seen in AlphaFold, these operations reason through every possible triangle of tokens (i, j, k) to enforce global geometric consistency. Boltz-2 addresses this challenge with a specialized GPU kernel known as TriFast[10]. By fusing multiple computational steps into a single, efficient GPU operation, TriFast achieves roughly a 2× speed-up over the standard PyTorch implementation.
In practical terms, this speed-up allows Boltz-2 to model larger molecular complexes without a slowdown, comfortably handling crops of up to 768 tokens at once. This boost in both depth and efficiency means Boltz-2 can capture more subtle molecular details and interactions—essential for accurately modeling large proteins, assemblies, and complex ligand-binding pockets.
The deeper, optimized Pairformer is now a powerful engine: it can model larger molecular scenes more accurately and do so faster. This is a key reason Boltz-2 can move confidently beyond static structure prediction, tackling the more challenging problem of predicting molecular function—like binding affinity.
Evolutionary Hints: Learning from the Family Tree
AlphaFold’s secret sauce has always been its use of evolutionary insights from multiple sequence alignments (MSAs). Boltz-1 inherited AlphaFold 3’s ability to optionally use MSAs as input, providing a valuable source of co-evolutionary information when available. Boltz-2 is designed to readily use MSAs, with its pipeline able to leverage external search tools to fetch evolutionary cousins, ensuring the model never flies blind when a family tree exists.
Moreover, Boltz-2 adds native support for paired MSAs. This is incredibly useful when two proteins in a complex, like a receptor and its binding partner, have evolved together across many species. By telling the model which sequences belong to the same organism, it can pay special attention to inter-protein co-evolution—the subtle, correlated mutations that occur between two different interacting proteins that have evolved in tandem across many species. This acts as a powerful tell-tale sign of where the two proteins have consistently touched throughout their shared evolutionary history.
Key Architectural Tweaks: From a Blueprint to a Refined Engine
Boltz-1 wasn’t just a straight copy of AlphaFold 3. Its creators acted like master mechanics, analyzing the original engine to find opportunities for improvement. They introduced several clever modifications aimed at enhancing information flow, training stability, and the reliability of predictions. Many of these thoughtful changes were carried forward into Boltz-2.
Fixing the Information Lag
Within AlphaFold’s trunk, the evolutionary data (MSA module) and the geometric blueprint (pair module) are in constant dialogue. The Boltz-1 team noticed a subtle quirk in this conversation: in each processing block, the geometric map was being updated using slightly “stale” information from the MSA’s previous state.
They realized it made more sense to use the freshest insights available. They reordered the operations so that each block now performs its updates in a more logical sequence:
- First, refine the understanding of the evolutionary data (the MSA representation).
- Then, immediately use that updated analysis to inform the geometric blueprint (the pair representation).
This simple change ensures the model’s structural hypotheses are always based on its most current evolutionary analysis. It’s like ensuring a detective always works with the latest clue, making the entire learning process more direct and logically consistent.
Building a More Stable Sculptor
AlphaFold 3’s generative core—the diffusion model that sculpts 3D coordinates from noise—is itself a specialized transformer. Deep transformers can be notoriously difficult to train; the learning signal (gradient) can fade as it travels back through many layers, a problem known as the “vanishing gradient.”
The Boltz-1 team made a critical and elegant change to fortify this sculptor: they added standard residual connections to its layers. Residual connections act like informational highways, allowing the input of a layer to skip past the main processing block and be added directly to the output. This guarantees that a smooth, uninterrupted gradient can flow back to the earliest layers, even in a very deep network.
By adding these connections, Boltz-1 dramatically improved the training stability of the diffusion model. It ensured that even the initial, chaotic steps of the denoising process received a strong learning signal, reducing the chances of the generative model getting “stuck” or failing to learn effectively.
The Rise and Fall of the “Mega-Judge”
A great model doesn’t just give answers; it knows when to be confident and when to be cautious. AlphaFold models predict their own accuracy using metrics like pLDDT and PAE. In AlphaFold 3, this was handled by a relatively small helper model [11].
The Boltz-1 team decided to transform this helper into an expert critic. They built a powerful, context-aware “mega-judge”—a 48-layer transformer as large as the main trunk itself. This judge was given a unique perspective: instead of just seeing the final predicted structure, it was shown a summary of the entire diffusion process. It got to watch the “movie” of the structure folding, from a random cloud of noise into its final form.
This trajectory awareness gave it remarkable nuance. It could spot subtle problems a normal judge might miss, like a flexible loop that was thrashing about unstably during generation. The result was a quality predictor more akin to an expert human who assesses not just the final answer, but the reasoning used to get there.
However, this powerful judge came at what the Boltz-2 paper describes as a “significant cost”. While Boltz-1’s large confidence model provided “some improvement,” the paper notes it is significantly more expensive than simpler architectures. In a pragmatic move for Boltz-2, the team opted for a faster architecture. They replaced the 48-layer mega-judge with a much more streamlined module using only eight PairFormer layers, which takes the final trunk representations and predicted coordinates as input but is no longer trajectory-aware.
It’s a classic engineering trade-off: a brilliant but complex system was simplified to create a robust and widely usable one.
With these core architectural improvements sorted, the team also had to tackle the monumental task of training the model, which required its own set of innovative data handling strategies.
Training a Titan: From Data to Discovery
Training a model as massive as Boltz-1 was a monumental undertaking, demanding innovative strategies to manage biological data far too vast for any single GPU. To accomplish this, the team developed several crucial techniques that transformed the model from theoretical architecture into a practical research powerhouse.
Unified Cropping Algorithm: Rather than applying spatial cropping (focusing on detailed 3D neighbors) and sequence cropping (taking continuous sequence segments) as separate steps, Boltz-1 merged both into a single, intelligent method. This unified cropping algorithm interpolates between the two extremes by randomly varying a “neighborhood size” for each training sample. As a result, the model experiences a wide range of structural contexts—from intricate binding-site details to broader, connected domain shapes.
Robust Pocket Conditioning:
To give researchers more control based on prior knowledge, Boltz-1 introduced robust pocket conditioning. This flexible feature allows the model to be guided even with partial information—a direct reflection of real-world scenarios, where complete binding site knowledge is rarely available. During training, the model is exposed to randomly selected subsets of known pocket residues, ensuring that it remains effective even when binding sites are only partially defined.
These sophisticated data-handling strategies were instrumental in efficiently training a robust, flexible model, laying the groundwork for Boltz-2’s next frontier: predicting not just structural outcomes, but functional insights.
The Diffusion Pipeline: Sculpting Structures with a Steady Hand
In Part 2 of this series, we saw how AlphaFold 3 swapped out its deterministic, piece-by-piece builder for a revolutionary new engine: a diffusion-based generative model. Boltz-1 and Boltz-2 embrace this same philosophy at their core, learning to sculpt a final, coherent structure out of a random, meaningless cloud of atoms.
Each of these “sculpting” steps is a sophisticated prediction. But the specific architectural choice for this generative model presented both a clever shortcut and a hidden trap.
The Engineer’s Dilemma: A Blessing and a Curse
One key design choice in both AlphaFold 3 and Boltz is that the diffusion model is not explicitly “equivariant.” In plain terms, the model isn’t hard-coded to know that rotating a molecule in space doesn’t change its internal structure. Instead, it must learn this concept by being shown thousands of examples in random orientations [12].
- The Blessing 🙏: This simplifies the network’s architecture. Using a standard, vanilla transformer is faster and easier to scale than using complex, specialized equivariant layers.
- The Curse ☠️: This engineering shortcut creates two subtle but serious problems: the “canonical pose loophole” and “rotational drift.”
Problem 1: The Canonical Pose Loophole
During training, after the model predicts a structure, an algorithm called Kabsch alignment rotates the prediction to best match the true answer before the error is calculated. This is meant to be fair—the model shouldn’t be punished for getting the overall orientation wrong. But this creates a loophole a “lazy” network can exploit.
A canonical pose is a single, fixed, “one-size-fits-all” orientation that the model might learn to output for every molecule, regardless of the input’s orientation. The training process can’t easily spot this cheat, because the Kabsch alignment step will always rotate the model’s lazy prediction to match the target, giving it a low error score. The model gets a good grade without ever learning the crucial skill of how to orient a structure correctly.
For a single protein chain, this might seem minor. But for a multi-protein complex, it’s a fatal flaw. If the model predicts two interacting proteins but places each in its own separate canonical pose, their relative orientation is completely random. The all-important interface between them is lost, and any predictions about their interaction are rendered meaningless.
Problem 2: The Rotational Drift Jitter
The second problem affects even well-behaved models. The diffusion process is iterative, taking hundreds of small denoising steps. At each step, the model’s prediction will inevitably have a tiny, almost unnoticeable rigid-body mismatch—a “jitter”—relative to the current noisy structure.
When you blend the prediction with the noisy cloud, this tiny rotational error introduces a small twist. Over hundreds of steps, these innocent-looking twists accumulate, causing the entire structure to drift in a slow spiral. This isn’t just visually unappealing; it can cause protein chains to become tangled or atoms to clash. It’s like a stop-motion animation filmed on a wobbly tripod—each individual frame’s movement is tiny, but when you play the movie, the character has drifted clear across the screen.
Boltz to the Rescue: Align-as-You-Go and Smarter Training
The Boltz team introduced two key improvements to solve these problems, transforming the diffusion pipeline into a more stable and predictable sculptor.
Solution 1: Align-as-You-Go Keeps the Chisel Steady
To prevent both the canonical pose cheat and the chaotic “rotational drift,” Boltz introduces Dynamic Kabsch Alignment at Every Step. In each reverse diffusion step, before the model’s prediction is blended with the current noisy structure, it is first perfectly aligned to it. This simple-sounding fix has profound consequences:
- It closes the loophole: By forcing the model to align to the correct frame at every single step, the “one-size-fits-all” canonical pose strategy becomes useless. The model has no choice but to learn the actual skill of orienting the structure correctly.
- It cancels out drift: The per-step alignment acts like tightening the wobbly tripod before every shot. It “re-zeros” the frame at each step, canceling out the small jitters before they can accumulate, ensuring the generation path stays smooth and direct, as shown in the right panel of Figure 2.
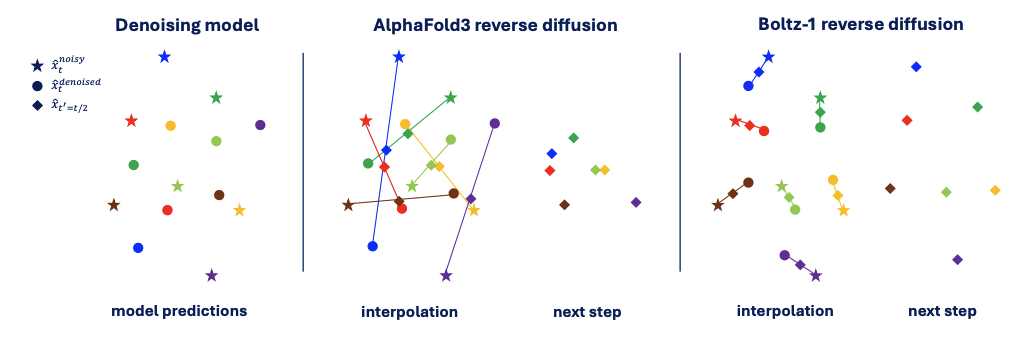 Figure 2. Align‑as‑you‑go keeps reverse diffusion stable. (a) Denoising network predicts a clean pose (stars) for noisy atoms xₜ (open circles); diamonds mark the half‑step target x̂ₜ′. (b) Vanilla AlphaFold 3 interpolates directly, producing long, divergent update vectors that twist the structure and accumulate rotational drift. (c) Boltz align‑as‑you‑go first rigid‑aligns the prediction to xₜ, then interpolates, giving short, collinear moves and gentler geometry updates.
Figure 2. Align‑as‑you‑go keeps reverse diffusion stable. (a) Denoising network predicts a clean pose (stars) for noisy atoms xₜ (open circles); diamonds mark the half‑step target x̂ₜ′. (b) Vanilla AlphaFold 3 interpolates directly, producing long, divergent update vectors that twist the structure and accumulate rotational drift. (c) Boltz align‑as‑you‑go first rigid‑aligns the prediction to xₜ, then interpolates, giving short, collinear moves and gentler geometry updates.
Solution 2: EDM Loss Weighting Trains a Versatile Artist
The team also borrowed a trick from cutting-edge image diffusion models. Instead of treating all stages of the generation process equally during training, they applied a clever EDM Loss Weighting strategy. This ensures the model becomes proficient across the entire spectrum of noise levels.
- High Noise (Early Stages): The training loss prioritizes making bold, imaginative leaps to establish the basic global shape.
- Low Noise (Late Stages): The loss prioritizes making fine, precise adjustments to perfect the local geometry and polish the final result.
The upshot is a more balanced and robust learning process, yielding a model that is adept at both creating a structure from scratch and refining near-perfect details.
Together, these improvements made the Boltz diffusion pipeline remarkably stable and predictable. With this solid foundation, the stage was set for Boltz-2 to tackle one of the biggest remaining criticisms of AI-generated structures: their physical realism.
With stability solved, Boltz‑2 could confront realism—chirality, clashes, and binding energy.
From Plausible to Physically Realistic: Boltz-2 Learns the Rules of Chemistry
One common critique of deep learning structure predictors is that they can sometimes produce models that look correct at a glance but have subtle chemical implausibilities. AlphaFold 3, for all its impressive accuracy, was not immune to this. For example, researchers found it could predict the wrong “handedness” (chirality) of drug molecules, or even get the stereochemistry of unnatural D-amino acids wrong over 50% of the time[13]—no better than a coin flip. Other issues included occasional steric clashes (atoms overlapping), unrealistic bond geometries, and warped planar groups.
Previous models tried to solve these issues with “after-the-fact” fixes, like using a penalty score to rank flawed models lower or running an energy minimization step to relax the structure. These were effective, but they felt like applying a band-aid rather than curing the underlying disease.
Boltz-2 addresses the problem at its core. It moves beyond structures that are merely plausible to ones that are physically realistic by integrating the rules of chemistry directly during the generation process. It introduces a system of Boltz-Steering potentials that act like a gentle hand on the tiller of the diffusion model, nudging it away from unphysical waters before it can get lost.
Boltz-Steering: Blending AI with Physics
When you enable Boltz-2’s physics-guided mode, the model activates a set of built-in potential functions that act as a “chemistry conscience.” These functions, inspired by classical molecular mechanics, are designed to gently guide the AI’s predictions toward physically sound conformations.
Boltz‑Steering operates at every reverse‑diffusion step. Here’s how it works:
At a given denoising step, the network makes its prediction for where the atoms should move. Simultaneously, the Boltz-Steering module evaluates the current structure against its chemistry rules and calculates a small correction. Think of it as a “guiding gradient” that points away from physical violations—for instance, away from a steric clash or toward a more ideal bond angle. This guiding gradient is then added to the AI’s own update, creating a final, combined move that is informed by both the network’s learned knowledge and fundamental physics.
Importantly, the strength of this physical guidance is applied in a time-dependent manner. The correction is almost zero at the beginning of the process when the structure is just a random cloud of noise—it doesn’t make sense to enforce chemistry on gibberish. As the structure takes shape and the noise recedes, the influence of the steering potentials ramps up, becoming strongest near the end of the generation process when the fine details like exact bond lengths and non-clashing side-chains matter most. This ensures the AI has freedom to make bold moves early on, while receiving precise, physics-aware guidance during the final polishing.
The key steering potentials teach the model a few core lessons:
- Steric Clash Avoidance: A gentle repulsive force, like an invisible spring, pushes any two non-bonded atoms apart if they get too close. This is crucial for cleaning up the crowded interfaces where molecules touch.
- Chain Entanglement Prevention: A mild “phantom force field” prevents separate protein chains from erroneously passing through each other, avoiding the dreaded “chain-mail jumble” that unconstrained models can sometimes produce.
- Bond Geometry Restraints: A set of harmonic (spring-like) potentials ensures that bond lengths and angles stay near their ideal, textbook values, preventing noticeable geometric distortions.
- Chirality and Planarity Enforcement: Perhaps most innovatively, Boltz-2 includes terms to preserve correct “handedness” and flatness. For each chiral center, a hefty penalty is applied if it tries to flip to its mirror image. For planar groups like aromatic rings, a flatness constraint counteracts any warping. These measures act as a crucial safety net, virtually eliminating the chirality errors seen in previous models.
For readers less versed in chemistry, here’s a quick intuition pump for the rules Boltz-2 enforces:
- Chirality (Handedness): Just like your left and right hands are non-superimposable mirror images, so are many molecules. Boltz-2 makes sure to keep the “handedness” correct—think of LEGO pieces that only fit one way.
- Planarity: Some molecular pieces are as flat as a playing card (like benzene rings). Boltz-2’s planarity check is like making sure all the “cards” in the molecular structure stay perfectly flat.
- Steric Clashes: Two objects can’t occupy the same space. The model now has a built-in “personal space” rule for every atom.
- Chain Entanglement: Think of two strands of cooked spaghetti. They can slide past each other, but shouldn’t pass through each other’s solid matter. Boltz-2 adds a force that prevents this from happening.
In short, Boltz-2 teaches the AI some basic manners of chemistry: keep your hands correct, your rings flat, and don’t step on each other’s toes.
It’s important to note that these potentials are applied softly. They guide, rather than dictate, allowing the model’s learned knowledge and the physics-based forces to find a harmonious balance.
New Levels of User Control: From AI Oracle to Interactive Partner
Beyond making the outputs more realistic, Boltz-2 also gives scientists more control over the modeling process, transforming it from a black-box oracle into an interactive research partner. Users can now easily inject their own expert knowledge into the prediction.
Key new control features include:
- Method Conditioning: A simple but clever feature where you can tell Boltz-2 what experimental context to assume with a flag (e.g.,
--method x-rayor--method md). This is like telling the model, “imagine this protein in a crystal lattice,” where it will favor a single, well-packed state, versus “imagine it flopping around in solution,” where it will allow for more flexibility. The full list of supported methods is extensive, including contexts like cryo-EM, solution nmr, and even theoretical model to handle various experimental and computational inputs[14] - Template Guidance: Boltz-2 supercharges the use of templates (known structures of related proteins). A scientist can provide a known protein structure as a template and ask the model to perform guided docking—folding and docking a new ligand while keeping the protein’s core structure tethered to the known conformation.
- Custom Distance Restraints: For advanced users, Boltz-2 accepts a simple input file with custom constraints. You can specify that two residues should be close based on experimental data, or that a disulfide bond must form. Each of these is translated into a gentle steering potential, allowing for a true human-AI collaboration in building complex structural models.
All these improvements—better architecture, stable diffusion, physical realism, and user control—make Boltz-2 a powerful platform for molecular design. But we haven’t yet touched on its headline feature: predicting the holy grail of binding affinity.
The Next Frontier: Predicting the Holy Grail of Binding Affinity
Perhaps the most revolutionary aspect of Boltz-2 is that it doesn’t stop at asking, “here is how these molecules might look when bound.” It goes further to ask the most critical question in drug discovery: “how strongly will they bind?”
This strength of interaction, or binding affinity, is what separates a potential blockbuster drug from a chemical dud. For decades, accurately predicting it has been the field’s holy grail. The gold-standard physics-based methods, like alchemical free-energy perturbation (FEP), are highly accurate but excruciatingly slow, taking days of supercomputer time for a single compound. This has limited their use to only a handful of the most promising, late-stage candidates.
Boltz-2’s affinity module shatters this paradigm. It aims to bring near-FEP accuracy to the earliest stages of discovery, providing rapid in silico affinity estimates in mere seconds. Let’s break down how this remarkable capability was engineered.
How the Affinity Module Works: The Inspector
After the diffusion model generates a plausible 3D structure for the complex, that structure is handed off to a specialized “inspector”—a separate computational head fine-tuned for a single purpose: evaluating interactions.
This affinity head is essentially a refined Pairformer that zooms in on the binding site with a magnifying glass. It takes the rich representations from the main trunk and scrutinizes the local region, judging the quality of the fit by analyzing every atomic contact, hydrogen bond, and hydrophobic patch.
This inspector then delivers a two-part verdict:
- A binary binding probability (
affinity_probability_binary): A simple 0-to-1 score answering, “Is this molecule likely to be a true binder, or is it a dud?” This is incredibly useful for virtual screening, allowing researchers to sift through hundreds of thousands of compounds to find the few that are actually worth pursuing. - A predicted affinity value (
affinity_pred_value): A numerical estimate of the binding strength, given as pIC50. This is the key metric for lead optimization, where chemists need to know if changing a molecule’s structure will make it bind ten times more tightly.
Training the Inspector: A Diet of Data and Deception
To build this inspector, the Boltz team needed to teach it the subtle language of physical chemistry. This required a monumental data engineering effort and several clever training strategies.
First, they compiled a massive dataset of over a million experimental binding measurements from public databases like ChEMBL and BindingDB. This included both quantitative values (like Kd and IC50) and qualitative, yes/no results from high-throughput screens.
Second, to ensure the model learned the right lessons, they used a crucial trick: synthetic decoys. For each known active drug, they found a similarly-sized molecule with a different shape and chemistry that was known to be inactive. By training on these look-alike pairs, the model was forced to learn the difference between true interaction patterns and superficial ligand properties.
Finally, they pushed the model further using distillation from its predecessor. Boltz-1 was used to generate thousands of extra training examples of plausible (but not always perfect) binding poses. Boltz-2’s affinity head was then trained to predict Boltz-1’s “opinion” on these structures, exposing it to a wider variety of poses and negative examples than static experimental data alone could provide.
The Payoff: Real-World Impact and Future Promise
The result of this intensive training is a model with a striking capability. On standard industry benchmarks, Boltz-2’s affinity predictions achieve a Pearson correlation of around 0.60–0.66 with experimental results. To put that in context, it’s in the same ballpark as some rigorous physics-based methods and far surpasses traditional docking scores. While not perfect, it represents a huge leap forward for AI in understanding the subtle energetics of binding.
It’s important to note the current caveats: the model only considers direct protein-ligand interactions (no explicit water molecules) and is not yet suited for protein-protein binding affinities. But as a proof of concept, Boltz-2 shows that a single neural network can master both structure and property prediction. It hints at a future where an AI can not just model a proposed drug, but also foretell its other properties, like toxicity or metabolism, all in one go.
Conclusion
The journey from AlphaFold 2 to the present day has unfolded like a three‑act play. First, AlphaFold 2 delivered a stunning solution to a 50‑year grand challenge, providing static, high‑accuracy structures for single proteins. Next, AlphaFold 3 made the generative leap, using diffusion models to predict the architecture of entire molecular assemblies. The third act has been an explosion of open‑source innovation, where models like Boltz democratized these powerful tools, refined their core engines, and pushed them toward answering deeper biological questions.
This wave of progress has fundamentally shifted the goals of the field. The frontier is moving beyond predicting static shapes and toward two new, more profound challenges. The first is capturing protein dynamics: understanding that proteins are not rigid objects but flexible machines that wiggle, breathe, and change shape. By training on data from molecular dynamics simulations, models like Boltz‑2 are beginning to capture this conformational diversity, predicting not just a single structure but a dynamic ensemble. The second frontier is predicting function. By directly estimating binding affinity, these new AI tools are asking not just “how does this drug fit?” but “how well does it work?”, a question that gets to the very heart of molecular efficacy.
The current landscape is no longer dominated by a single player but is a vibrant ecosystem of open tools. While Boltz has pushed the envelope on affinity, neighboring projects—RoseTTAFold All‑Atom, ESMFold, Bio‑Emu, and the antibody‑centric Chai‑1—are exploring their own essential niches. The open nature of these models means their powerful base architectures can be readily adapted or fine‑tuned for specialized biological problems. This is a virtuous cycle: each open model lowers the barrier for the next, turning what began as a singular breakthrough into a community‑driven engine of innovation.
With human insight and AI now working side by side to co‑design dynamic molecular machines, faster drug discovery and deeper biological understanding feel less like distant goals and more like the next stop on an open road.
-
Sengar, A. Deconstructing the Fold with AlphaFold 2. Blog Post, 2025. link ↩
-
Sengar, A. Denoising the Complex with AlphaFold 3. Blog Post, 2025. link ↩ ↩2
-
AlphaFold 3 – Launched as a web‑only tool in May 2024; full source + weights became downloadable on 11 Nov 2024 under the Creative Commons CC‑BY‑NC‑SA 4.0 licence, which allows research but forbids commercial use. Boltz‑1 – Released in Nov 2024 under the MIT licence with model weights, making it the first AF3‑class system that is completely free for any purpose, including commercial R & D.Chai‑1 – Open‑sourced in Oct 2024 under Apache‑2.0 focused on antibody and small‑peptide tasks rather than general protein‑ligand complexes. ↩
-
Wohlwend, J., Corso, G., et al. (2024). Boltz-1: Democratizing Biomolecular Interaction Modeling. bioRxiv preprint 2024.11.19.624167 link ↩
-
Passaro, S., Corso, G., Wohlwend, J., et al. (2025). Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv preprint 2025.06.14.659707. link ↩ ↩2
-
FEP is a cornerstone of computational chemistry, providing highly accurate predictions of binding affinity by rigorously simulating the physical transformation of one molecule into another. This accuracy, however, comes at an immense computational cost, which has historically limited its use to only a handful of the most promising drug candidates. ↩
-
Bio-IT World (2025, June 6). MIT Researchers Unveil Boltz-2: AI Model Predicts Protein Structure, Binding Affinity in Seconds. link ↩
-
Abramson, J., Adler, J., Dunger, J., et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(7930):493–500, 2024. ↩
-
RDKit is a widely-used open-source cheminformatics toolkit. In this context, it generates plausible 3D conformers for small molecules from their 2D chemical structures. ↩
-
TriFast is a “fused kernel,” meaning it reduces multiple computational operations into a single, efficient GPU call. This minimizes memory overhead and significantly accelerates computation. NVIDIA’s cuEquivariance library[15] achieves even greater kernel-level optimizations (up to 5×), underscoring the growing importance of hardware-software co-design in modern deep learning. ↩
-
In AlphaFold 3 this job fell to a lightweight “confidence head” — an 8‑layer mini‑PairFormer that reads the trunk’s final features to output pLDDT and PAE. ↩
-
This stands in contrast to AlphaFold 2’s Structure Module, which used complex, explicitly equivariant attention (IPA) to avoid this issue entirely. ↩
-
Childs, H., Zhou, P., & Donald, B. R. (2025). Has AlphaFold 3 Solved the Protein Folding Problem for D-Peptides?. bioRxiv, 2025-03. link ↩
-
NVIDIA Developer Blog. Accelerated Molecular Modeling with NVIDIA cuEquivariance and NVIDIA NIM microservices. link ↩