
I work on generative models for all-atom protein motion that learn from molecular dynamics and structure data. At EPFL, I built LD-FPG: a spectral GNN autoencoder + latent diffusion system that samples physically plausible backbone and side-chain ensembles for GPCRs, with open code and a public D2R dataset. I’m extending this with latent-space dynamics—score-guided Langevin, Koopman, and neural autoregressive propagators—to recover kinetics, transition paths, and pathway usage for design. My earlier works span oxDNA-based DNA nanotechnology (branch migration, mismatch proofreading, free-energy mapping) and multiscale catalysis: a particle-based simulator linking surface chemistry to reactor performance, including studies of catalyst deactivation and transport effects. Across projects I prioritize open datasets, reproducible code, and methods that connect physics with machine learning.
- My Time at EPFL (2024 - current)
- My time at Imperial College London — DNA Nanotechnology & Biomolecular Systems (2020-2024)
- PhD days (2015-2020)
- Misc
My Time at EPFL (2024 - current)
Developing a Generative AI Framework for All-Atom Protein Dynamics (Accepted at NeurIPS 2025)
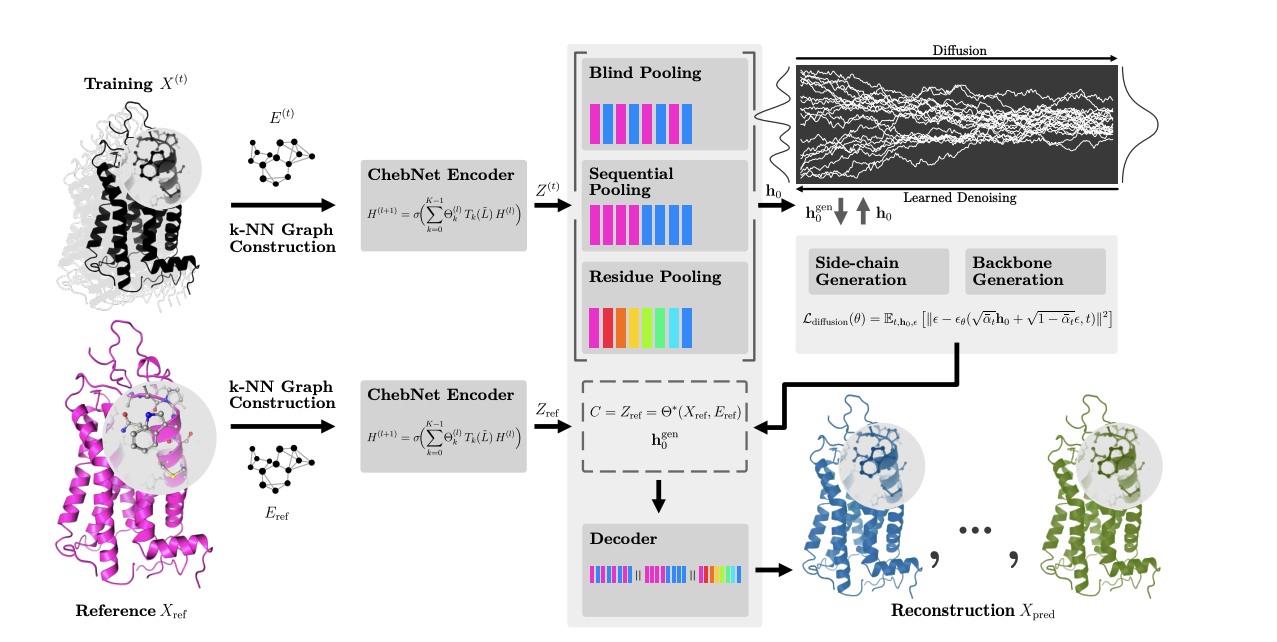
A major challenge in computational biology is capturing the full range of motion of proteins, as their function is tightly linked to their dynamics. To address this, I developed Latent Diffusion for Full Protein Generation (LD-FPG). My contribution centred on designing and implementing the machine-learning pipeline: a spectral graph neural network autoencoder coupled to a latent-diffusion model. I also validated the method on a complex, medically relevant target — the human dopamine D2 receptor (D2R) — showing that LD-FPG can generate complete, all-atom conformational ensembles directly from molecular-dynamics data.
Our framework reproduced both the global backbone architecture and the distributions of side-chain dihedral angles — dynamics essential for molecular recognition. This work provides an efficient tool to study the dynamics of challenging proteins and opens new avenues for structure-based drug design. In the spirit of open science, I curated and released a full D2R molecular-dynamics dataset along with the complete LD-FPG codebase to encourage further innovation.
Video demonstration of LD-FPG model (EPFL work)
To illustrate the power of our Latent Diffusion for Full Protein Generation framework, below are two clips from our trained models. The first shows raw molecular-dynamics (MD) snapshots of the human PKC-δ C1 domain followed by frames generated by LD-FPG. You can see how the model captures backbone flexibility, helix breathing and loop motions. The second clip shows another protein (3dAN). Once trained, LD-FPG can generate thousands of physically realistic, all-atom conformations in seconds — ideal for ensemble docking, AI-augmented scoring and any workflow requiring a rich representation of protein motion.
Resources
[1] Latent Diffusion for Full Protein Generation (LD-FPG) — (NeurIPS 2025) preprint (arXiv: 2506.17064).
[2] LD-FPG code — GitHub repository.
[3] All-atom MD dataset (D2R) — Zenodo record 10.5281/zenodo.15479781.
Beyond Ensembles: Simulating All-Atom Protein Dynamics in a Learned Latent Space (Accepted at NeurIPS AI4Science)
TL;DR. We fix the LD-FPG encoder–decoder and swap only the latent propagator to study what truly drives long-horizon rollouts. We compare three options—score-guided Langevin, a Koopman linear operator, and a neural autoregressive model—on ADP → 7JFL → A_1AR and recover the A_2AR activation surface. The neural model is most stable over long horizons; Langevin gives the sharpest side-chain rotamers; Koopman is an interpretable, lightweight baseline.
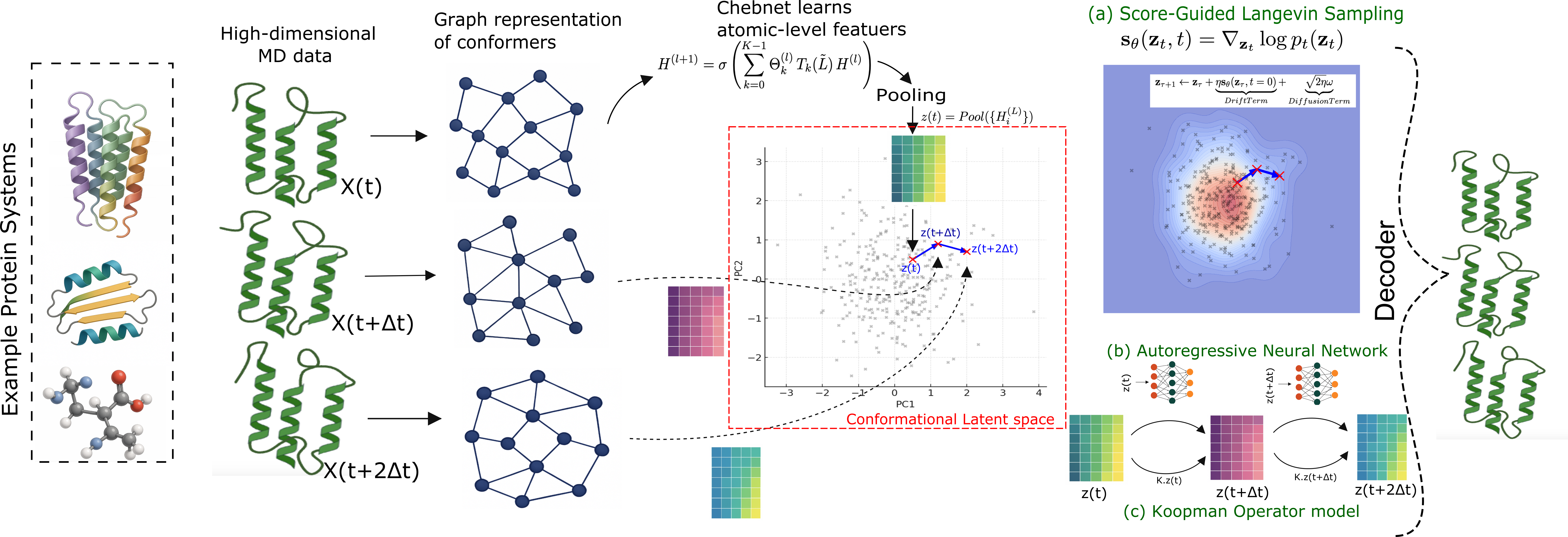
Key takeaways
- Controlled comparison: same latent, same decoder; only the propagator changes.
- Backbone vs side-chains: autoregressive > backbone fidelity; Langevin > side-chain rotamers.
- Scaling: works from small peptides to GPCRs; reproduces A_2AR activation surface.
Resources
- Paper: Beyond Ensembles: Simulating All-Atom Protein Dynamics in a Learned Latent Space — arXiv: 2509.02196
- Code: GLDP — github.com/adityasengar/GLDP
Rollout clips
Computationally Reprogramming GPCR Allosteric Pathways
I co-authored a new study introducing a structure- and dynamics-driven design approach that infers and rewires allosteric signal flow in GPCRs, enabling ligand-selective reprogramming of receptor responses. The method maps communication pathways across the 7TM scaffold and proposes mutations that redirect information flow toward targeted effector routes. In head-to-head tests across multiple ligands, designed variants shifted pathway usage and reshaped signaling preferences, highlighting a path-centric route to bias control and drug design.
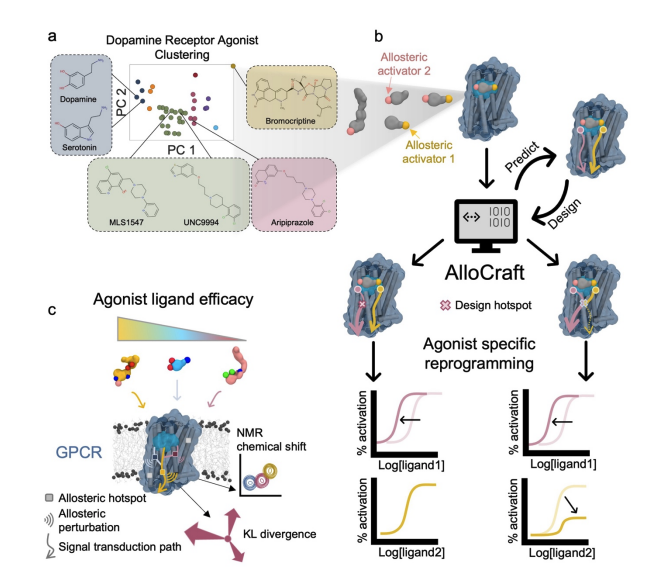
Resources
| [NEW] Computational design of allosteric pathways reprograms ligand-selective GPCR signaling — bioRxiv preprint (2025). PDF | Landing page |
My time at Imperial College London — DNA Nanotechnology & Biomolecular Systems (2020-2024)
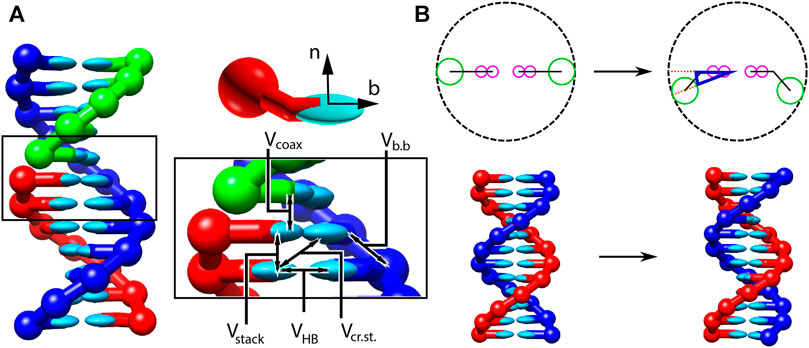
I combined method development, simulations and theoretical analysis to advance DNA nanotechnology using the oxDNA framework. This work, showcased at multiple international conferences and through several posters, tackled key challenges in DNA design and function.
1. Free-energy mapping of four-way DNA junctions
Using enhanced sampling techniques based on oxDNA, I mapped the free-energy profile of four-way DNA junctions — crucial intermediates in strand-displacement reactions. Surprisingly, introducing bulges actually destabilizes the junction due to entropy penalties. This finding challenges common assumptions and provides guidance for designing more robust DNA nanostructures.
2. Kinetic proofreading in nonenzymatic DNA strand displacement
I applied oxDNA to model a kinetic proofreading (KP) mechanism in nonenzymatic strand-displacement systems. The study provided quantitative reaction-rate estimates and showed that operating under out-of-equilibrium conditions dramatically enhances recognition of single-nucleotide mismatches. These insights inform highly specific applications such as single-nucleotide polymorphism detection and DNA-based diagnostics.
3. oxDNA primer: when to use it, how to simulate, how to interpret
We produced a detailed primer on the oxDNA coarse-grained model. The article explains model variants (oxDNA1/oxDNA2), force-field details, sequence-dependent parameterization and mapping to experimental units. It also walks through simulation protocols — Langevin dynamics, Monte Carlo and advanced accelerated sampling methods such as Virtual Move Monte Carlo (VMMC) — demonstrating how these can speed up equilibration of large, strongly interacting DNA structures. Worked examples show how VMMC explores conformations of DNA origami and multi-strand assemblies and combines with umbrella sampling to obtain free-energy profiles. The primer also details analysis workflows for computing structural observables, thermodynamic quantities and reaction pathways, providing a benchmark reference for reproducible oxDNA studies.
Additional method
I developed a fast-kinetics sampling workflow in oxDNA to study dynamic events such as DNA bubble formation; this approach has since been adopted by groups at the University of Cambridge and MIT (submitted).
Resources
[4] Overcoming the speed limit of four-way DNA branch migration with bulges in toeholds — Nano Letters (2025). 10.1021/acs.nanolett.5c03063. Preprint: bioRxiv 10.1101/2023.05.15.540824. Dataset & analysis code: Zenodo 10.5281/zenodo.15398317.
[5] Kinetic Proofreading Can Enhance Specificity in a Nonenzymatic DNA Strand Displacement Network — Journal of the American Chemical Society (2024). DOI: 10.1021/jacs.3c14673. Open access: PubMed Central. Dataset: Zenodo 10.5281/zenodo.8132461.
[6] A Primer on the oxDNA Model of DNA: When to Use it, How to Simulate it and How to Interpret the Results — Frontiers in Molecular Biosciences (2021). DOI: 10.3389/fmolb.2021.693710. Open access: PMC. Example files: Zenodo 10.5281/zenodo.4809769.
PhD days (2015-2020)
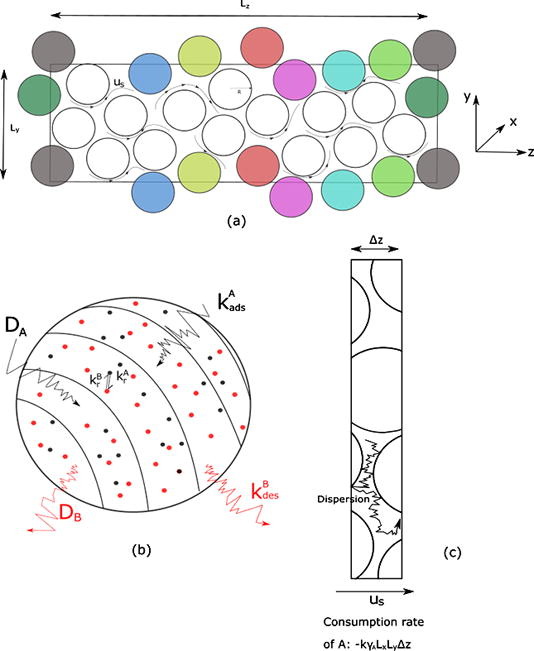

My research has addressed a core barrier in chemical engineering: connecting molecular-level surface events to macroscopic reactor performance. I developed a particle-based computational-fluid-dynamics framework that provides a unified, multiscale view of catalytic processes. The core innovation was resolving small-scale physics — diffusion and surface reactions — while seamlessly operating at the scale of industrial reactors. The model was designed and validated to switch between reaction-, diffusion- and convection-dominated regimes, a versatility previously out of reach for real-time simulations. I further advanced this platform by incorporating complex, nonlinear surface kinetics, enabling accurate and scalable simulations of multicomponent systems and their mass-transfer fluxes under nonequilibrium conditions.
Building on this platform, I applied the model to a pressing industrial problem: catalyst deactivation in sustainable aviation-fuel production. I developed and compared two theoretical models that connect molecular-scale deactivation mechanisms with observable reactor-scale performance degradation. By analyzing industrial alkylation-reaction data, I identified a previously unknown molecular compound that acts as a potent deactivating agent. The models also revealed that the deactivation rate is highly sensitive to proton mobility on the catalyst surface. This analysis offers a strategy for extending catalyst lifetime by optimizing proton interactions, providing a clear path to reduce downtime and improve the economic viability of sustainable fuel production.
Collectively, this work constitutes a complete “molecule-to-reactor” predictive pipeline, demonstrating a progression from method development to high-impact industrial application. In recognition of its novelty, the multiscale model [8] was featured on the cover of Chemical Engineering Science (Vol. 198), and the deactivation research earned me an invitation to present at Faraday Discussions, a leading international forum for groundbreaking research.
Resources
[7] Particle-based modeling of heterogeneous chemical kinetics including mass transfer — Physical Review E (2017). DOI: 10.1103/PhysRevE.96.022115.
[8] Towards a particle-based approach for multiscale modeling of heterogeneous catalytic reactors — Chemical Engineering Science (2019). DOI: 10.1016/j.ces.2018.10.038.
[9] Deactivation Kinetics of Solid Acid Catalyst with Laterally Interacting Protons — ACS Catalysis (2018). DOI: 10.1021/acscatal.8b01511.
[10] Deactivation Kinetics of the Catalytic Alkylation Reaction — ACS Catalysis (2020). DOI: 10.1021/acscatal.0c00932.
[11] The challenge of catalyst prediction — Faraday Discussions (2018). DOI: 10.1039/C7FD00208D.
Misc
Residence Time in Asymmetric Simple Exclusion Dynamics
I proposed this study on particle transport, modeling how particles move through a crowded 2D strip using an Asymmetric Simple Exclusion Process (ASEP). We used Mean Field and Birth-and-Death models to predict the particle residence time, comparing these theories to Monte Carlo simulations. While our models perfectly captured positive-drift dynamics, we found a complex, non-monotonic behavior in the zero-drift “bouncing back” regime. Here, a small amount of horizontal diffusion surprisingly reduces residence time by allowing particles to bypass jams.
Resources
[12] Residence time estimates for asymmetric simple exclusion dynamics on strips — Physica A: Statistical Mechanics and its Applications (2016). DOI: 10.1016/j.physa.2015.09.037.
Blockchain Technology for Efficient Public-Sector Schemes
I proposed using blockchain as a distributed ledger for securely storing and managing beneficiary data, with smart contracts automating transactions to reduce human intervention. We applied this framework to India’s flagship employment scheme, MGNREGA, which had an allocation of roughly ₹55,000 crore (~US $8 billion) in FY 2018–2019. By leveraging cryptographic integrity and immutability, the system ensures funds are disbursed only to verified recipients and that all transactions are auditable, improving accountability and reducing fraud and inefficiency.
Resources
[13] Blockchain technology in efficient implementation of Government-based schemes — preprint (ResearchGate link) — View.